
Last Updated: 1/26/2023
Thank you to the PRISMS Professional Advisory Board for putting together this FAQ; to Laura S. Schmidt, Ph.D. at the National Cancer Institute for her expertise and additional review; and to SMS parent Jill Effron for championing this effort.
What is Birt-Hogg-Dubé syndrome and how is it related to SMS?
Birt-Hogg-Dubé syndrome (BHD) is an adult-onset medical condition characterized by symptoms involving the skin, lungs, and kidneys. It is a rare genetic disorder caused by changes (mutations) in a single gene called folliculin (FLCN). FLCN happens to be located within the 17p11.2 chromosomal region that is deleted in most people with SMS. Although most adults with SMS do not develop BHD, it is important for families and professionals to be aware of the connection between these two separate disorders.
What are the symptoms of BHD and when do they occur?
The primary symptoms of BHD involve the skin, lungs, and kidneys. The estimated rates of occurrence for each symptom and ages of onset are based on studies of individuals and families diagnosed with BHD. Since these studies did not include people with SMS, the actual risk of BHD symptoms in individuals with SMS is unknown but may be significantly lower than described in families with FLCN mutations.
Skin:
Small dome-shaped bumps (known as fibrofolliculomas, also trichodiscomas) appear on the face, neck, and upper body in most adults diagnosed with BHD over the age of 25. These skin changes are generally not dangerous to one’s health but may create cosmetic concerns. Fibrofolliculomas must be confirmed by biopsy, as skin tags and bumps are common in the general population and are usually not related to BHD.
Lungs:
Most people with BHD develop cysts in their lungs after age 30. By themselves, these cysts rarely cause noticeable symptoms. However, these cysts are associated with an increased risk for a more serious condition called spontaneous pneumothorax (also known as collapsed lung). By age 40, about 30% of adults with BHD will have had one or more episodes of pneumothorax. Onset is typically in adulthood but can occasionally occur in children. Pneumothorax is a painful and potentially serious condition that requires urgent medical treatment.
Kidneys:
Up to one third of older adults with BHD develop kidney cancer (renal cell carcinoma), usually in their late 40s to early 50s, but occasionally in young adulthood. The type of kidney cancer that develops most often in BHD patients tends to be less aggressive and less likely to spread throughout the body (metastasize) than other types of kidney cancer, so there are fewer reported cases of cancer-related deaths among BHD patients than in other types of kidney cancer. Individuals diagnosed with BHD require routine MRI scans of the abdomen to check for signs of cancer so that treatment can be started early.
How do mutations in FLCN cause BHD, and are adults with SMS at risk?
Scientists believe that BHD occurs due to a “two-hit” process. Most individuals diagnosed with BHD are born with a mutation in the genetic code of one copy of their two FLCN genes. This first mutation is present in every cell in the body, but by itself is not enough to cause the clinical symptoms of BHD. However, over the course of a person’s lifetime, a random change in one or more cells in a person’s second copy of FLCN (a second hit) can trigger BHD. This second mutation typically occurs in lung, skin, or kidney cells, resulting in the features of the disorder. Those in whom a second mutation occurs will be more likely to develop BHD manifestations, whereas other people born with one FLCN mutation may never develop a second mutation and will not develop BHD. However, the risk to develop a second mutation is a lifetime risk.
FLCN is one of dozens of genes located within the 17p11.2 chromosomal region. Most people with SMS are born with a 17p11.2 deletion that includes FLCN. As with mutations, one deleted copy of FLCN is not enough to cause BHD clinical symptoms. However, as they age, adults with SMS are theoretically at risk for developing BHD if the copy of FLCN on their non-deleted chromosome 17 happens to randomly undergo a mutation in some of their cells. In other words, because of their 17p11.2 deletion, most people with SMS are born with a first hit, putting them at risk for developing BHD if the FLCN gene on their non-deleted chromosome 17 receives a second hit.
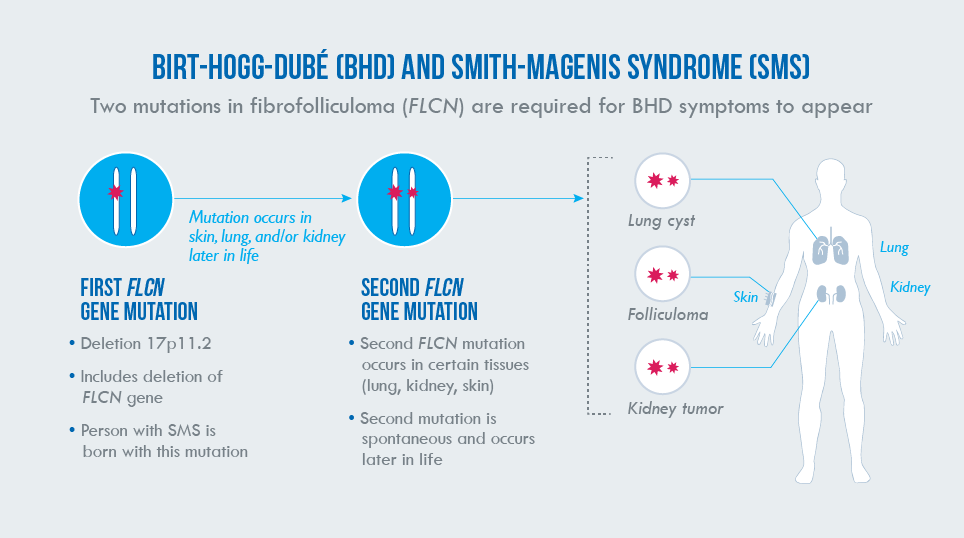
Individuals with SMS due to RAI1 mutations and those with uncommon deletions that do not include the FLCN gene are at no greater risk than the general population for developing BHD. There has been theoretical concern for increased renal cancer risk in patients with SMS since the majority of SMS cases result from a 3.7 Mb deletion in 17p11.2, which includes the FLCN gene. In 2014, Ann C. M. Smith and her colleagues at the NIH described two adults with SMS (50-year-old woman & 45-year-old man) with kidney cancer; they have since identified two additional unrelated adults with SMS and co-occurrence of BHD features leading to their recent publication (Vocke et al., 2023). Among these four cases, three constitute the first known association of histopathologically verified characteristic BHD-associated renal tumors in adults with SMS; the fourth was identified to have histologically confirmed skin fibrofolliculomas.
What now?
Since BHD is considered an adult-onset disorder, we do not recommend any additional screening in children with SMS for symptoms related to BHD. Importantly, the diagnosis of BHD is based on the presence of specific features discussed previously. Most people with SMS have an increased chance of developing BHD, but the diagnosis should never be made in children or adults who show no evidence of features. Having one hit (an SMS deletion) does not mean that a person has or ever will develop the clinical features of BHD.
Learn more about BHD
In its most recently published GeneReview of SMS, the PRISMS Professional Advisory Board includes the following recommendation for people with SMS who have a confirmed diagnosis of BHD:
“The common deletion results in haploinsufficiency (one missing copy) of FLCN that is associated with Birt-Hogg-Dubé (BHD) syndrome, raising a theoretic concern for increased risk of renal carcinoma (kidney cancer) in individuals with SMS… While unstudied, the co-occurrence of renal tumors in a few unrelated adults with SMS suggests that precautionary cancer surveillance may be considered in adulthood for individuals with co-occurring BHD syndrome.”
Translated, this means that adults with SMS and their families should be aware of BHD symptoms and discuss any concerns with their healthcare providers. If an adult with SMS is diagnosed with BHD, the usual guidelines for BHD monitoring should be considered, including regular abdominal imaging using MRI for early detection of cancer. Current recommendations suggest abdominal imaging of kidneys with initial screening at age 20, with repeat lifelong screening every 3 years.
Although many studies have been published on both BHD and SMS as separate disorders, little is known about how these two disorders interact together. More research remains to be done, and PRISMS’ professional advisors will continue to monitor new developments. As always, participation by PRISMS families in the SMS Patient Registry and SMS research will be crucial to finding answers.