Below is a summary of the research findings of PRISMS funded post-doctoral fellow, Takao Tsukahara D.D.S Ph.D. Takao was funded by PRISMS from January 2018 to March 2020. He will be continuing his work on RAI1 at the University of Michigan. We are excited to share his current findings with our community, and look forward to his future research!
Takao Tsukahara D.D.S Ph.D
Drs. Iwase and Sutton Laboratories
University of Michigan
Summary
Previous studies revealed that Rai1 is involved in gene expression. However, the role of Rai1 in the gene expression program evoked by environmental stimuli is poorly understood. Given that adaptive behavior and synaptic plasticity require evoked gene expression and these function are impaired in many neurodevelopmental disorders which share common features with Smith Magenis syndrome (SMS), it is thus important to understand Rai1’s role in the gene expression program driven by environmental stimuli.
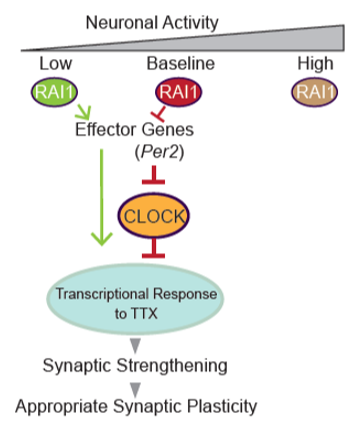
So far we found that Rai1 regulates at least two distinct gene expression program. Firstly, Rai1 regulates the gene expression program that maintain steady-state condition in neural network upon normal activity by optimizing synaptic strength. Secondary, Rai1 regulates the gene expression program driven by reduced network activity and increase synaptic strength. This function contributes to bring back the network activity in an optimal range where neurons could function properly.
With these findings, we noticed that the top misregulated genes upon Rai1 reduction were those involved in the sleep-wake cycle. This finding led us to ask if the aberrant expression of sleep-wake cycle genes are leading to observed defects in synaptic plasticity. If so, interventions in these genes might ameliorate not only the sleep-wake abnormalities but also refine other brain functions in SMS patients. Therefore, now we are manipulating the expression of Per2, a sleep-wake cycle related gene which is increased in Rai1 reduced culture, and ask if this could restore the observed phenotype.
Method
RNA-seq
Mouse cortical neural cultures were infected with either non-targeting control sh-RNA or sh-Rai1 to reduce Rai1. After 3-days of infection, cultures were treated with Na+ channel blocker tetrodotoxin (TTX; 1 µM, 24 hrs) or the GABAA receptor antagonist bicuculline (Bic; 20 µM, 24 hrs), respectively, to drive the activity-dependent gene expression program. Next, to detect the very small change in gene expression, we chemically labeled the nascent RNA and conducted RNA-seq.
Electrophysiology
We co-transfected shRNA-expression plasmids with a GFP-expression plasmid in primary hippocampal neurons (12-14 days in vitro, DIV), which targets roughly 1% of neurons allowing for an analysis of cell-autonomous roles of Rai1. We then induced synaptic upscaling or downscaling through the application of the Na+ channel blocker tetrodotoxin (TTX; 1 µM, 24 hrs) or the GABAA receptor antagonist bicuculline (Bic; 20 µM, 24 hrs), respectively.
Results
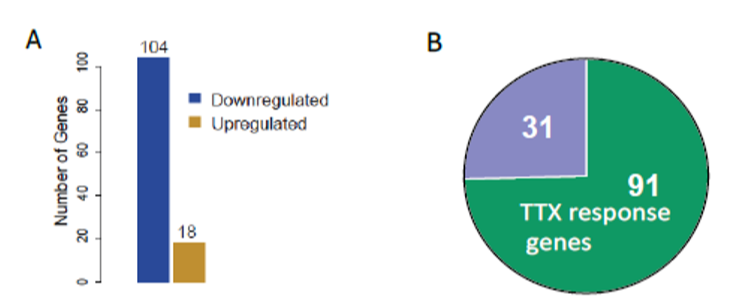
RNA-seq enabled us to profile the gene expression programs upon change in network activity. Then, we classified the misregulated genes in Rai1-KD cultures at baseline. Surprisingly, most of the misregulated genes upon Rai1-KD were those responding to TTX, reduced network activity (Fig.1).
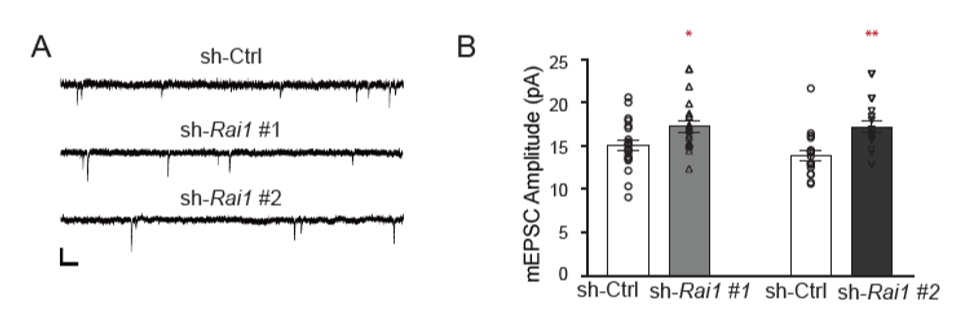
Next, since chronic network silencing induce up scaling and Rai1 reduction evoked TTX response genes, we recorded miniature-excitatory post synaptic currents (mEPSC) to ask if Rai1-reduction results in increased synaptic strength. Indeed, we found an upward shift of synaptic strength upon Rai1 reduction. We found an upward baseline shift in the mEPSC amplitude of RAI1-KD neurons (Fig. 2) .
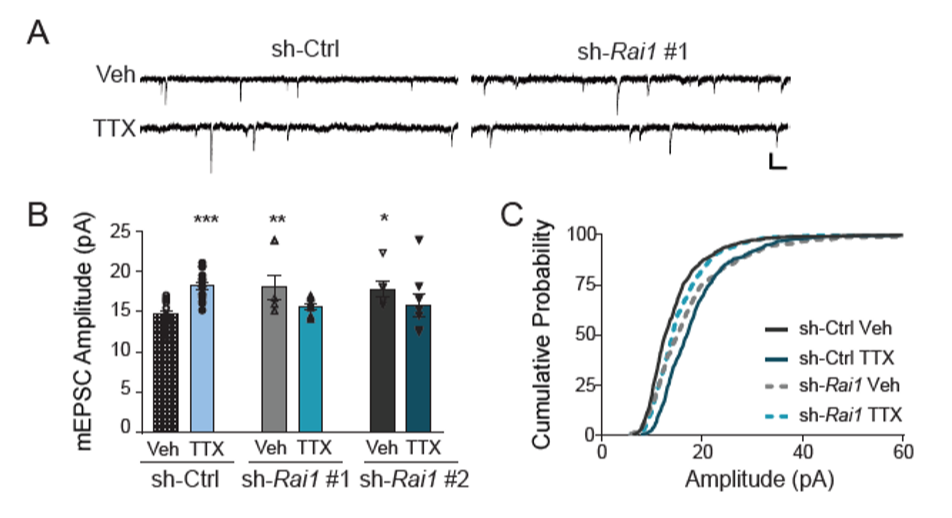
In addition, we found that the Rai1 reduced cultures fail to evoke the gene expression program driven by reduced activity (data not shown). Then, we asked if synaptic up scaling, which requires the gene expression program driven by reduced activity, will also be impaired in Rai1 reduced neurons. Consistent with the RNA-seq data, we saw impairment of up-scaling upon Rai1 reduction (Fig. 3).
Future directions
During this characterization of Rai1, we noticed that Rai1 reduction leads to aberrant expression of sleep-wake cycle genes. Therefore, we are asking if manipulating the expression of sleep-wake cycle genes such as Per2 could refine the abnormal synaptic strength, restore appropriate synaptic plasticity and lead to proper neural functioning (Fig. 4).